 |

Automatic sequencing error correction
About sequencing errors and contig ambiguities
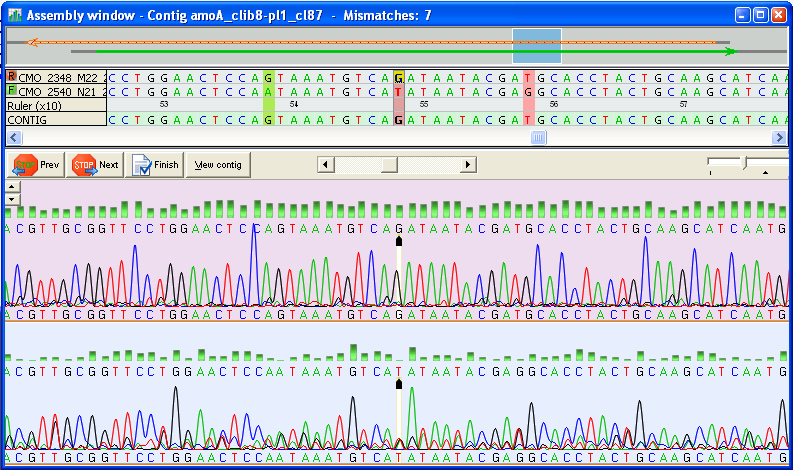
There are cases when the assembled chromatograms contain overlapping peaks. The base caller software in the sequencing machine usually resolve these peaks poorly, resulting in ambiguous (IUPAC) or even erroneous bases. Fortunately, the base caller recognizes that there might be problems with those peaks and therefore it assigns them low QV (confidence scores).
Automatic contig ambiguity correction
- When the confidence score is not available
Contig ambiguities can be corrected when two or more sequences are overlapping in the assembly process, since it is very likely that at least one sequence will contain a high QV (correctly identified) base. When calculating the contig, DNA Baser Assembler chooses the base with the higher QV (confidence scores). Therefore, when performing sequence assembly, DNA Baser Assembler is also correcting the ambiguities resulted from sequencing errors. The corrections suggested by this algorithm are near perfect. Cases where ambiguity correction algorithm cannot precisely suggest the correct base are extremely rare!
ADVANTAGES: Because of DNA Baser's precise ambiguity correction algorithm you don't need to open each contig and manually inspect/edit it. Once the assembly process is done, you simply move on and assemble the next pair of sequences.
- When the confidence score is not available
The automatic ambiguity correction algorithm kicks in only when the confidence score info is available in ALL input chromatograms. If you are mixing chromatogram files and plain text files (FASTA, TXT or SEQ) the ambiguities in the contig are corrected using a different ambiguity correction algorithm, since the plain text files cannot store information about confidence scores.
This algorithm chooses the majority base (for example, if for the same position the bases are C, T, T and G, the T will be chosen as the correct base). This is less accurate but still good enough to correctly fix the ambiguities in 99% of the cases.
Note that in some cases (when the software in the sequencing machine was missconfigured) not even the ABI/SCF files will contain the ambiguity scores. In this case we recommend you to use DNA Sequence Assembler's integrated base caller. This module is available since v4.0. More info about our base caller here. We recommend using our integrated base caller even if your chromatogram files already contain QV info since the tests shown that values computed with our base caller are more accurate that the ones computed with classic programs such as Phred.
Manual contig ambiguity correction
The user's hands-on time is minimized by our AMBIGUITY NAVIGATION SYSTEM, that guides the user from ambiguity to ambiguity and allows fast editing of the incorrect base. For more details about the NAVIGATION SYSTEM, click here.
Details about the contig ambiguity correction algorithm
When deciding between two or more bases, the algorithm will chose whichever has the highest confidence score. However, there are cases when the algorithm has to decide between a base and a gap. Since gaps do not have assigned any confidence score, the choice is entirely based on the confidence score of the base. If the confidence score is higher than a preset confidence score threshold (default 25, can be changed by the user), then the base is chosen. However, if the confidence score of the base is lower than the confidence score threshold, then the gap is chosen. For more details about setting the confidence score threshold, click here.
|
|