| |
|
|
BATCH SEQUENCE ASSEMBLY BY NAME PATTERN
In order to assemble samples at batch, DNA Baser needs to detect which files should be assembled together. Therefore, your samples must be named in a recognizable pattern based on their membership to a contig. In Batch Assembly Parameters tab (see figure below), you can define the characteristics of your name pattern.
How to use it?
Example 1a: Let's suppose we have a set consisting of two files:
The characters that are common (invariables) for both files are "EColli7", while the character that variates is "F" and "R". Therefore, we use the mouse to select the invariable part: EColli7. Now the program will know that EColli7F and EColli7R belong to the same contig.
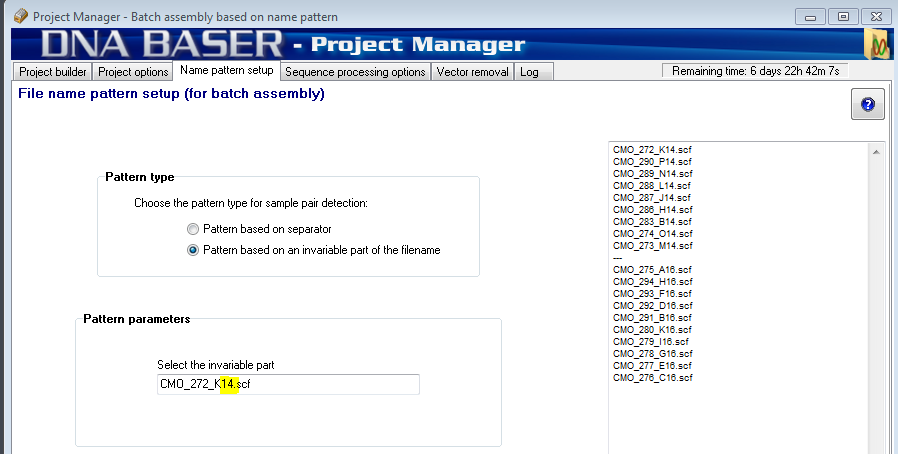
Example 1b: Let's suppose we have two sets. Each set has two files: Set1:
Set2:
We see that the first set has an invariable part: 14. The second set has also an invariable part: 16. We use the mouse to select those two characters that are invariable (the program will show only the first sequence from your Job List):
Example 2 (using separator): Let''s suppose we have a set consisting of two files with variable name length:
The first part of the name does not follow a pattern so it is useless. However, the characters after the underscore ("00A") are common (invariables) for both files Therefore, enter the underscore in the 'Separator' box and tell the program that 'The second part of the name is invariable'.
TUTORIAL Assemble thousands of samples in minutes!
Scenario
You have a clone library of 500 sequences and you use two primers (Forward and Reverse) to sequence each clone. At the end of sequencing process, you will have a folder with 1000 sequences, which need to be assembled in 500 contigs. It would be rather tedious to assemble a contig at a time. However, DNA Baser is the only software that allows you to assemble all sequences in one-step. The prerequisite is that the sequences that belong to the same contig are: a. Named after a pattern OR b. Placed in a separate folder (in this case see Batch sequence assembly by sub-folders)
Let's start
Start DNA Baser. The Project Manager should open by default. Prepare DNA Baser for sequence assembly (you need to perform this step only once).
Select the samples to be assembled
Add all samples that you want to assemble into the JOB LIST.
Set the name pattern
DNA Baser detects the pairs of samples based on the name of the sequences. Please visit this page to see how you should set the pattern.
Start the assembly
To start the assembly, just press the START BATCH ASSEMBLY
Job complete!
During the batch assembly process, a detailed log is generated. It contains information about each individual assembling process, a batch job summary, the list of parameters used for assembling, quality of each assembly job and many many other statistics.
During batch assembly, DNA Baser creates the following folders (located in current folder):
Output
Unassembled
Unpaired
Related topics:
|
||
| SciVance Technologies |