 |
NextGen Sequence Workbench
A graphic FastQ/SFF viewer & editor
The way bioinformatics programs should be...
NextGen Sequence Workbench is an efficient and easy to use FastQ/SFF file viewer, editor and converter. NextGen Sequence Workbench is the first and only complete FastQ/SFF editor with graphic interface on the entire bioinformatics market! With this tool we wanted to help the biologists to concentrate on their work instead of wasting time poking commands in an obscure text console.
The program is able to open huge FastQ/SFF files even when running o a modest computer.
Why is it special?
- SFF and FastQ support in one single unified graphic user interface
- Faster and lighter that FastQC
- Monolithic & portable (requires no installation, works under guest/limited accounts)
- Requires no additional add-ons (Java, .Net, etc)
- Supper efficient memory usage. Opens any file no matter how large.
- Free
Features
FastQ encoding auto detection
- SFF
- FastQ, FQ
- Auto-detect encoding
- Sanger encoding
- Solexa encoding
- Illumina 1.0, Illumina 1.3, Illumina 1.5, Illumina 1.8 encoding
- FastQ encoding auto detection
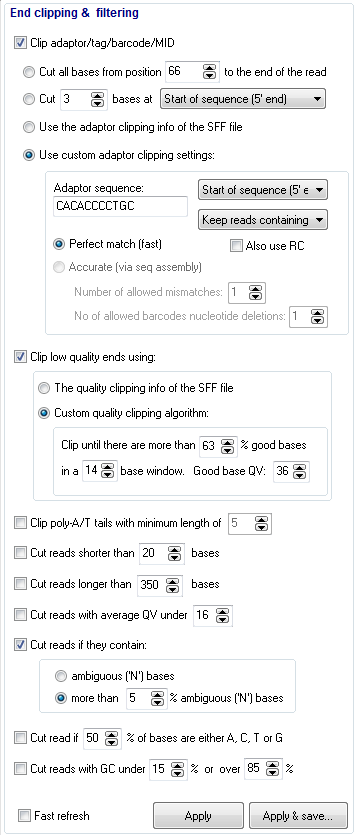
Filters
- Cut poly-A/T tails
- Cut reads with average QV under specified threshold
- Cut reads if they contain N bases (the user can specify how many)
- Cut low complexity reads
- Cut reads that are too short
- Cut reads that are too long
- Cut low quality ends. Automatically detect and cut low quality bases at the end of each read
Tools and converters
- Split multiplexed files (MID/Barcode splitter)
- Remove contaminants (search over represented sequences against a contaminant database)
- Dereplicate sequences (under development)
- File splitter: Split huge FastQ/SFF file in chunks of x reads
- File splitter: Cut all sequences in the specified range
- Compact FastQ files
- Convert SFF to FastQ
- Convert SFF to Fasta
- Convert FastQ to Fasta (multiFasta)
- Convert FastQ file to a different encoding (under development)
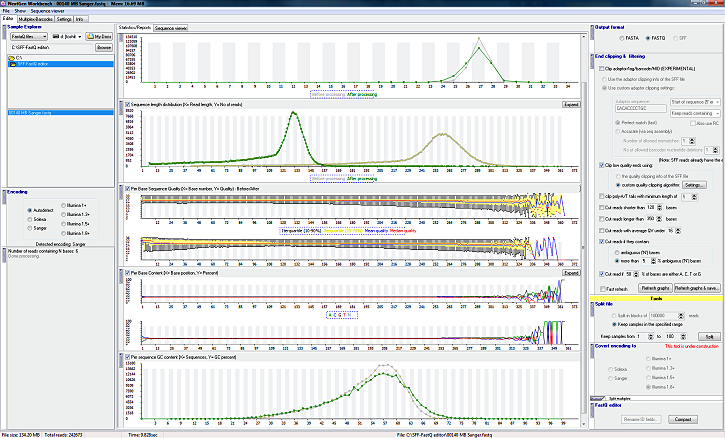
Graphs and data analysis
- Read lister - Show read name, base sequence, average quality, sequence length
- Graphic quality representation for each individual read. Color coded.
- Sequence length distribution graph
- Per base sequence quality graph
- Per base GC content graph
- Per base sequence content graph
- Per base N content graph (integrated in the 'Per Base Content' graph)
- Per sequence quality scores graph graph
- Duplication level graph
- Overrepresented sequences table
- Graphs can be expanded to full screen
- All graphs are updated in real time as the file is processed!
- File statistics: file name, file size, number of reads (before and after filtering), encoding, per file GC percent
- The analysis of the current file (including all graphs) can be saved as HTML report
GUI / System
- Easy to use/configurable GUI
- Save/remember GUI state
- Open SFF/FastQ files when user double clicks them in Windows Explorer
- Open file passed via command line
- Easy to use file browser (allows you to quickly locate your FastQ/SFF files)
- Long operations can be easily canceled
|
Related tools:
- NCBI BLAST DB Downloader is a a freeware biology software tool that automates the NCBI BLAST DB download process.
- NCBI Blaster (aka BLAST Robot) is a software tool that automates the NCBI BLAST search processes.
|
|
|
Download
NextGen Sequence Workbench is now part of the Avalanche NextGen package.
| NextGen Sequence Workbench |
| Version |
3.2.3 |
| Date |
August 2015 |
| Download time |
less than 10 seconds |
| DOWNLOAD |
|
Requirements
- Few MB of disk space
- < 30MB free RAM
- No Java, no .Net
Portability
This software tool is really small so you can easily copy it on a floppy disk or USB flash stick and take it with you or send it to your colleagues via email. |
Feedback
This tool is  . Please let us know how to make it better. . Please let us know how to make it better.
We are interested in:
- Feature requests
- Platform you are interested in (Windows, Mac, Linux)
- Statistics about your files (file type, how many, file size) and your working station (CPU/RAM)
- A name for the program :)
Performance
NextGen Sequence Workbench processes a 500MB file in under 10sec. The memory footprint never exceeds 15-30MB. Details
|


{kind=link}